FLEXIBLE WAVEFUNCTION METHODS FOR STRONGLY CORRELATED SYSTEMS
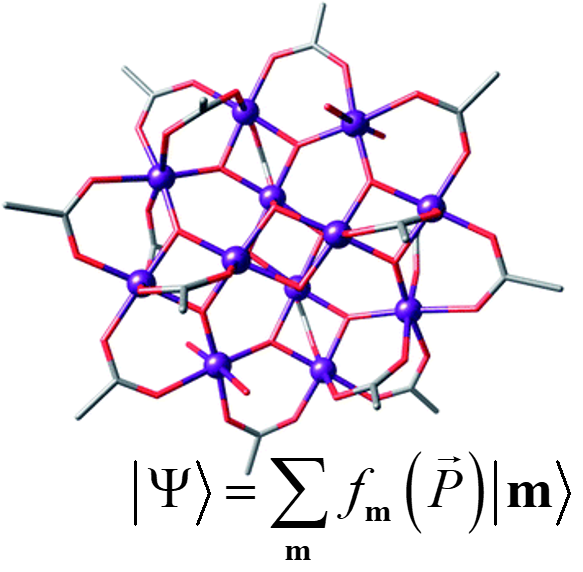 Accurately solving the electronic Schrödinger equation provides many enticing possibilities, from understanding a wealth of chemical phenomena, to facilitating the design of compounds with desired characteristics. Unfortunately, there are many cases for which current theoretical methods are either too computationally costly or lack sufficient accuracy. This is particularly evident in strongly correlated systems (e.g., systems with many unpaired electrons, or where bonds are stretched or broken). We are developing novel wavefunction methods to study these systems using a generalization of the Configuration Interaction (CI) method: the Flexible Ansatz for N-body Configuration Interaction (FANCI). Our main focus at the moment is on Coupled Cluster (CC) and quasi-particle (e.g., geminal) wavefunctions, but we are also looking forward to considering more “exotic” ansätze. We are also working on flexible perturbation theory strategies, efficient ways to include dynamic correlation corrections, and new wavefunction-in-wavefunction embedding techniques.
Accurately solving the electronic Schrödinger equation provides many enticing possibilities, from understanding a wealth of chemical phenomena, to facilitating the design of compounds with desired characteristics. Unfortunately, there are many cases for which current theoretical methods are either too computationally costly or lack sufficient accuracy. This is particularly evident in strongly correlated systems (e.g., systems with many unpaired electrons, or where bonds are stretched or broken). We are developing novel wavefunction methods to study these systems using a generalization of the Configuration Interaction (CI) method: the Flexible Ansatz for N-body Configuration Interaction (FANCI). Our main focus at the moment is on Coupled Cluster (CC) and quasi-particle (e.g., geminal) wavefunctions, but we are also looking forward to considering more “exotic” ansätze. We are also working on flexible perturbation theory strategies, efficient ways to include dynamic correlation corrections, and new wavefunction-in-wavefunction embedding techniques.
Representative publications:
https://arxiv.org/abs/2102.01171
https://arxiv.org/abs/2102.01224
CHARGE TRANSFER AND CHEMICAL REACTIVITY
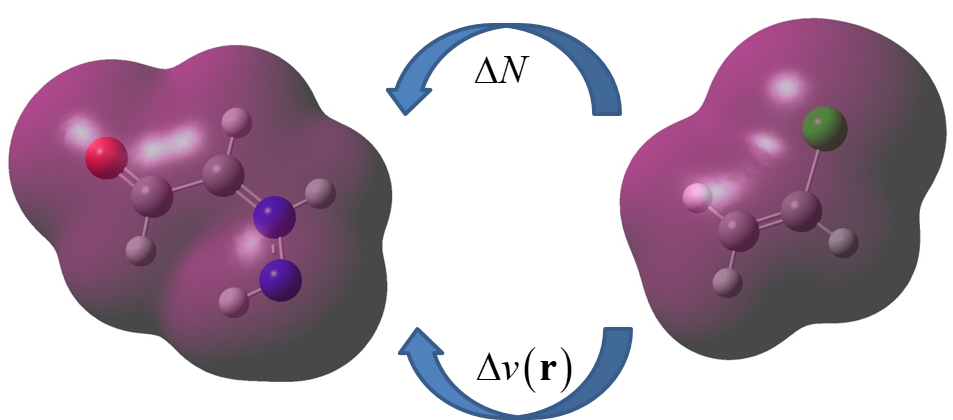
We are working on realistic models to accurately describe how electrons are transferred in chemical processes. A central theme of our research is analyzing how the energy of a system changes in response to a variation in its number of electrons. We have pioneered a framework that allows us to consider how the environment perturbations modify the charge-transfer properties of a system. We are now expanding this formalism and applying it to the study of chemical reactivity, atomistic models, redox reactions, and solvation processes.
Representative publications:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7756232/
https://www.sciencedirect.com/science/article/pii/S0167732220348479
CHEMICAL DIVERSITY AND DATA ANALYSIS
 Quantifying the “distance” between objects is a central theme in many research areas. This is particularly relevant in chemistry, where the old dictum that “similar compounds have similar properties” has proven pivotal in drug and materials design. We are working on algorithms that allow us to calculate the similarity of any number of molecules with unprecedented efficiency. We are using these tools to find new ways to navigate the chemical space and to develop novel methodologies for drug design. We are also applying our framework to process imaging mass spectrometry (IMS) data, clustering molecular dynamics trajectories, and extracting information from DNA and protein databases.
Quantifying the “distance” between objects is a central theme in many research areas. This is particularly relevant in chemistry, where the old dictum that “similar compounds have similar properties” has proven pivotal in drug and materials design. We are working on algorithms that allow us to calculate the similarity of any number of molecules with unprecedented efficiency. We are using these tools to find new ways to navigate the chemical space and to develop novel methodologies for drug design. We are also applying our framework to process imaging mass spectrometry (IMS) data, clustering molecular dynamics trajectories, and extracting information from DNA and protein databases.
Representative publications:
https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00505-3
https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00504-4